Dan Koboldt
Since the completion of the Human Genome Project in 2001, we've uncovered a great deal about the so-called Book of Life. The completed (draft) sequence of the human genome represents one of the foremost scientific achievements of our time. It also raised just as many questions as it did answers. Perhaps the most-debated of these was simply, where did all the genes go?
Disappearing Genes
In the 1990s, around when the HGP was first getting under way, scientists estimated that our genome might contain as many as 100,000 protein-coding genes. They derived this number from early surveys of “expressed sequences” (RNA transcripts) isolated from different organs and tissues. Over time, as technologies improved, the estimated number of human genes moved ever downward. In 2000, most estimates were below 50,000. A year later, the HGP published its landmark findings, which included their prediction of up to 40,000 protein-coding genes.
After three years of detailed “finishing” work, a much-improved draft of the human genome was released in 2004. With it came a new estimate: 20,000 to 25,000 protein-coding genes. Now, in 2016, the most popular view favors an estimate at the low end: 19,950 protein-coding genes.
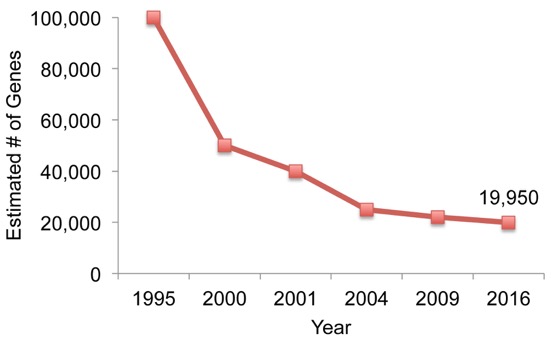
Figure 1. Number of predicted human protein-coding genes by year, 1995-2016.
Adapted from M. Pertea and S.L. Salzberg, Genome Biology 2010;11(5):206..
This modest number of genes surprised many scientists, particularly because we’ve sequenced the genomes of many other organisms that have more than 20,000 genes. As Pertea and Salzberg wrote in a wonderful 2007 review, the current gene count puts humans “between chicken and grape.”
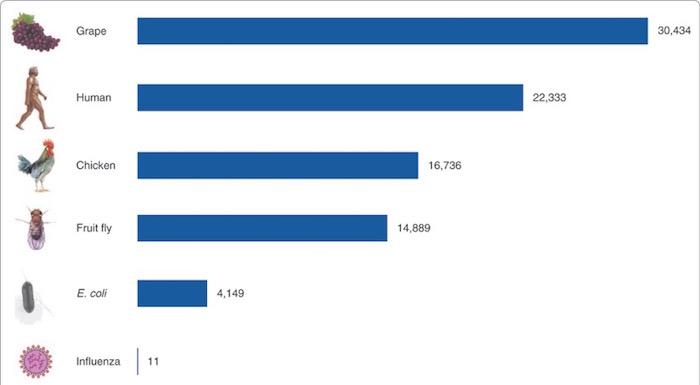
Figure 2. Gene counts in a variety of species (M. Pertea and S.L. Salzberg, Genome Biology 2010;11(5):206.).
Although the number of genes surprised many, the completion of the HGP shed light on a deeper and more troubling fact: the human genome encompasses 3.2 billion base pairs—equivalent to 1,600 copies of J.R.R. Tolkien’s The Lord of the Rings trilogy—and scientists had little idea what most of it does.
The Genome’s Dark Matter
Around 35 million bases in our genome encode known proteins. That seems like a lot, but remember, the genome itself is 3.2 billion base pairs. All of the coding bases combined represent around 1% of the total. We certainly don’t understand the functions of all human genes, but at a minimum, we can say that the function of coding bases is to make proteins. The function of the other 99%—the “dark matter” of the human genome—is less obvious.
We know that some of it is functional, too. If you compare the sequence of the human genome to that of other vertebrates (mouse, rat, bird, etc), around 5-10% of the sequences are “evolutionarily conserved,” meaning that they’re >90% identical in species that diverged from one another millions of years ago. This suggests that they serve some vital purpose; otherwise, the process of random mutation and genetic drift would have altered them long ago.
In 2004, researchers at the University of California, Santa Cruz, reported almost 500 regions of our genome that were 100% identical between human, mouse, and rat. Many of them were millions of base pairs long, and located in regions of the genome that don’t have any known protein-coding genes. For three years, there was a lot of excitement (in the research community, at least) about these “ultra-conserved elements” and what their vital function might be. In 2007, however, a follow-up study found that gene knockouts of four ultra-conserved elements in mice still produced viable offspring that were otherwise healthy.
Nevertheless, the fact that evolutionary pressures have maintained many noncoding sequences in distantly related species suggests that they play an important role. This, along with a surprisingly modest number of human protein-coding genes, would suggest that noncoding parts of the genome regulate the coding part, thereby giving rise to the complex organism that we claim to be. For the last fifteen years, genomic scientists have worked to understand the nature of that regulation. Shedding light on the dark matter, as it were.
Regulatory Elements
In 2003, the NIH funded an ambitious effort to identify regulatory elements of the genome and figure out what they do. The ENCyclopedia Of DNA Elements (ENCODE) project consortium used a variety of new genomic technologies to interrogate DNA from various tissue types, with the goal of capturing regulatory machinery in action. They sequenced RNA transcripts (an indicator of which parts of the genome are being transcribed), searched for DNA-protein interactions, measured “epigenetic” alterations, like methylation of cytosine, etc.
So what have ENCODE and other research groups uncovered in the dark matter of the human genome? All kinds of interesting things. Perhaps the most famous of these was the phenomenon of widespread transcription, in which RNA polymerase enzymes copy a DNA sequence into its complementary RNA form. In high school biology, you probably learned about three types of RNA:
- Ribosomal RNA (rRNA), which aids in protein synthesis as a component of ribosomes, A.K.A. the protein factories of a cell.
- Transfer RNA (tRNA), which transports amino acids to the ribosome so that they can be incorporated into proteins.
- Messenger RNA (mRNA), which is transcribed in the nucleus and provides the template for building those proteins.
Previously, scientists expected that most of the RNA molecules in a cell were messenger RNAs that originated from genes. Surprisingly, the ENCODE project revealed that transcription was happening all over the place. Around 70% of the human genome produces RNA at some point in some cell type. Most of these RNAs are not classic ribosomal, transfer, or messenger RNAs, but likely perform a different kind of regulatory function.
MicroRNAs, for example, are short (18-24 nucleotide) sequences that participate in a process called RNA silencing. The first of these was discovered in the 1990s, but it took years for us to recognize their importance in cell function. MicroRNA genes encode a somewhat longer precursor molecule of about 80-100 nucleotides with a palindromic sequence that causes it to fold back on itself in a hairpinlike structure. These are recognized and cleaved by Argonaute proteins, which use the microRNA sequence to attach to complementary sequences in messenger RNAs to prevent their translation into amino acids. In other words, microRNAs “silence” specific mRNAs before they can make proteins.
The behavior of microRNAs helped explain a strange phenomenon called RNA interference, in which the injection of double-stranded RNA into a cell causes suppression of messenger RNAs that have a corresponding sequence. It only takes a few molecules of double-stranded RNA to trigger complete suppression of the target mRNA, no matter how many copies the cell makes. Furthermore, the suppressive effect can spread between cells. RNA interference represented a new type of regulation in the cell (post-transcriptional regulation), and the discovery of its underlying mechanism won the Nobel prize in 2006.
Other types of regulatory “dark matter” are not transcribed, but perform their function by binding to specific proteins. In the promoter of most genes, for example, is a certain base sequence (TATAAA) that serves as the binding site for RNA polymerase II—the enzyme that makes messenger RNA from DNA. Noncoding regions also contain binding sites for other regulatory players: transcription factors, which control gene activity, and splicing factors, which ensure that messenger RNA is processed correctly.
Many regions of the genome bind to histone proteins, which package and order DNA so that it can fit into a relatively small space (the nucleus) in an organized fashion. Think of histones as spools around which you wrap a bunch of thread (DNA) to keep it nice and orderly. This packaged DNA is called chromatin, and it generally has two states.
- Heterochromatin, which is tightly packed, inaccessible to regulatory proteins, and thus not actively producing transcripts.
- Euchromatin, which is more loosely packed and where most of the action happens.
Heterochromatin is essentially the default state. Open chromatin (euchromatin) requires proteins to bind and protect it from being wrapped up into heterochromatin. Undoubtedly, many noncoding regions of the genome serve to bind these protective proteins, thereby ensuring that nearby genes can be activated as necessary.
The ENCODE Project and other efforts have identified thousands of large genomic regions that actively regulate protein-coding genes. Enhancer regions, for example, are big stretches of noncoding DNA that help drive the activity of certain genes. These regions are believed to have binding sites for transcription factors and other proteins. Often, they are near the genes whose activity they enhance, but they can also be located thousands of base pairs away. Repressors are elements that do the opposite: they prevent genes from being transcribed. Usually, this is accomplished by recruiting proteins that either bind or make chemical modifications to DNA so that it's inaccessible to the transcription machinery.
Structural Elements
Of course, not all of the genome’s 3.2 billion bases can be vitally important. Like the old imperial starships that Rey salvages in Star Wars: The Force Awakens, our genome contains a lot of junk. Around two-thirds of it is highly repetitive, serving little known purpose for biology. In fairness, some of these repetitive regions exist simply as a buffer for more important regions.
For example, at both ends of our chromosomes are special structures called telomeres. These are stretches of a six-letter sequence (TTAGGG, in humans) repeated over and over again. They're essentially disposable bases that shorten over time due to a quirk of DNA replication. At birth, our telomeres are about 11,000 bases long; by old age, they’ve shortened to around 4,000 bases. This gradual shortening occurs because DNA replication machinery can’t copy all the way to the end. It needs a place to hold on, similar to how the slider can’t open the last few teeth on a zipper, without falling off the zipper.
Thus, chromosomes become a little bit shorter every time they’re copied for cell division. The telomeric repeats ensure that they don’t lose anything important. Similarly, there’s a repetitive structure in the middle of the chromosome where sister chromatids (the original chromosome and its freshly-made copy) are connected during cell division. Again, that’s not a place where you want to store the vital genes.
Conclusion
In summary, our genome is a huge, complex thing that we don’t completely understand. The parts that we’ve studied the most—the protein-coding regions—represent just 1% of it. Yet our initial forays into studying the noncoding portions, the “dark matter” of the genome, have shown that they play important roles as well. As technologies continue to improve, we’ll unlock even more secrets about the fundamental code of life.
The only consistent fact about our genome is that the more we study it, the more complicated it becomes. This calls for caution when we consider altering the human genome, which has become increasingly feasible with the development of new genetic engineering technologies like CRISPR/Cas9. Within a decade, I think it’ll be possible to correct a disease-causing mutation in a human embryo to prevent a future genetic disease. But I expect it will be a much longer time—a century, perhaps, before we can engineering a genetically superior human like they do in Gattaca. And once we can, we’ll have to think about whether or not we really should.
Copyright © 2016 Dan Koboldt
Dan Koboldt is a genetics researcher with more than 60 publications in Nature, Science, The New England Journal of Medicine, and other journals. He works at an NIH-funded genome sequencing center, where he and his colleagues use next-generation DNA sequencing technologies to uncover the genetic basis of inherited diseases such as age-related macular degeneration, heart disease, cancer, and diabetes. Dan also writes science fiction and fantasy. His debut novel The Rogue Retrieval, about a Vegas magician who infiltrates a pristine medieval world, is out from Harper Voyager. His web site can be found here.